| نام آزمایش: | شارکوت مارى توث |
|---|---|
| نمونه مورد نیاز: | 5cc خون وریدی |
| مدراک مورد نیاز: | کلیه آزمایشات مربوطه و دفترچه درمانی |
| نحوه نمونه گیری: | مراجعه حضوری و دریافت 5cc از خون وریدی فرد |
| شرح از تست: | نوروپاتی ارثی شارکوت ماری توث (CMT) که تحت نام نوروپاتی حسی حرکتی ارثی(Hereditary Motor and Sensory Neuropathy (HMSN , بیماری وراثتی اعصاب حسی و حرکتی Hereditary Sensorimotor Neuropathy (HSMN) و همچنین آتروفی عضلانی ساق پایی Peroneal Muscular Atrophy شناخته میشود بیماری بسیار هتروژن است که اعصاب محیطی را درگیر میکند. نام شارکوت ماری توث از نام سه متخصص اعصاب گرفته شده که برای اولین بار در قرن نوزدهم این بیماری را توصیف کردند. در این بیماری سیستم عصبی حسی و حرکتی با اختلال مواجه میشود که ناشی از به هم خوردن تدریجی ساختار و عملکرد اعصاب محیطی (نوروپاتی) است. این اختلال به صورت متقارن در اندامها به ویژه پاها و در مراحل پیشرفته دستها بروز میکند. شارکوت ماری توث با میزان شیوعی در حدود 1 در هر 33 هزار نفر شایع ترین نورپاتیارثی است. نشانه های بیماری معمولاً در اولین تا سومین دهه زندگی فرد به صورت ضعف و آتروفی عضلانی آشکار میشود. در بعضی منابع بیماری را بدون درد توصیف کردهاند. اما این نوروپاتی میتواند با درد هم همراه باشد. کف پای قوسی شکل و انگشتان چکش مانند از نشانه های این بیماری است. جهش در ژنهایی که در رشد و نمو اعصاب محیطی نقش دارند باعث بروز این بیماری میشود. الگوی توارثی این ژنها متفاوت است. بر این اساس این بیماری را تقسیم بندی میکنند. تقسیم بندی این ژنها چندان ساده نیست چون در مواردی دیده شده که موتاسیونهای مختلف در یک ژن باعث بروز بیماری با الگوی توارثی متفاوتی میشود. هر کدام از بیماریهای ذکر شده در جدول یک خود بر اساس ژن آسیب دیده و محصول پروتئینی ایجاد شده به چندین بیماری تقسیم میشود. شارکوت ماری توث نوع یک (CMT1): این نوع از بیماری شارکوت ماری توث با آسیب پوشش میلینی در اعصاب محیطی مشخص میشود. سستی و آتروفی عضلانی، از دست دادن حس و پایین آمدن سرعت هدایت اعصاب تا حد 5 تا 30 متر در ثانیه از نشانههای بیماری است ( در حالت نرمال این میزان بیشتر از 40 تا 45 متر در ثانیه است). پیشرفت بیماری به صورت تدریجی است و علائم بیماری بین 5 تا 25 سالگی آشکار میشود. کمتر از 5 درصد بیماران وابسته به ویلچر میشوند. بیماری معمولاً باعث کوتاه شدن دوره زندگی نمیشود. 6 نوع CMT1 وجود دارد که از لحاظ تظاهرات بالینی مشابه هستند و تنها با آزمایش ژنتیک از هم تفکیک داده میشوند. ژن درگیر و موتاسیون ایجاد شده در زیر گروههای این بیماری در جدول 2 مشخص شده است .  شارکوت ماری توث نوع 2 (CMT2): در این نوع CMT سلولهای اکسونی اعصاب محیطی با اختلال مواجه میشوند. از لحاظ بالینی مشابه نوع 1 است اما کاهش هدایت جریان عصبی در این نوع کمتر است. CMT2 پانزده زیر گروه دارد که نشانههای بالینی آنها مشابه است و با آزمایش ژنتیک از هم تفریق داده میشوند. اگرچه ژن درگیر در بیشتر این زیر گروهها شناخته شده است اما در مواردی نیز تنها کروموزم حامل این ژن شناسایی شده و بنابراین تست ملکولی مربوط به آن هنوز به صورت آزمایشگاهی وجود ندارد (جدول3). فراوانی زیر گروههای CMT2 نیز هنوز شناخته نشده است.  شارکوت ماری توث نوع حد وسط: در این نوع از شارکوت ماری توث تظاهرات فنوتیپی مربوط به اکسونها و سلولهای میلین غیر عادی مشاهده میشود. سرعت هدایت جریان عصبی از CMT1 و CMT2 بیشتر است. الگوی توارثی این بیماری اتوزومال غالب است. آزمایش تشخیص این نوع CMT در حال حاضر تنها به صورت تحقیقاتی انجام میشود.  شارکوت ماری توث نوع 3 و 4 (CMT3 و CMT4): در بعضی منابع شارکوت ماری توث نوع 3 و 4 را هر دو با هم تحت نام شارکوت ماری توث نوع 4 با توارث اتوزومال مغلوب دسته بندی میکنند. در این دسته بندی شارکوت ماری توث نوع 4 را با اکسونوپاتی و میلینوپاتی در نظر میگیرند. زیر گروههای CMT4 بر اساس تفاوتهای کلینیکی، آسیب شناختی و ملکولی از یکدیگر تفریق میشوند. جدول 4 انواع این بیماری را نشان میدهد. تشخیص اکثر این بیماریها به صورت معمول در آزمایشگاهها انجام میگیرد. تشخیص انواعی از آنها نیز محدود به آزمایشات تحقیقاتی است. 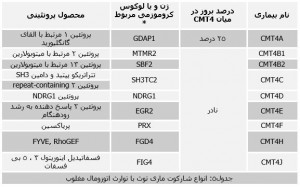 شارکوت ماری توث وابسته به کروموزم x: نوروپایی شدید تا متوسط حسی و حرکتی که معمولاً مردان را مبتلا میکند و در زنان ناقل علامتی ندارد. این رده از بیماری شارکوت ماری توث به پنج زیر گروه تقسیم میشود. CMTX2 و CMTX4 علاوه بر نوروپاتی با عقب ماندگی ذهنی همراه است. از دیگر تظاهرات بالینی که در CMTX4 و CMTX5 دیده میشود ناشنوایی است.  از میان انواع مختلف این بیماری در این آزمایشگاه تشخیص سه نوع شارکوت ماری توث نوع 1A (CMT1A) ، شارکوت ماری توث نوع 2A2 (CMT2A2) و شارکوت ماری توث نوع (CMT4A ) 4A انجام میگیرد. شارکوت ماری توث نوع 1A شارکوت ماری توث نوع 1A در اثر دوپلیکاسیون در ژن PMP22 رخ میدهد. این بیماری با نامهای مختلفی چون Hereditary Motor and Sensory Neuropathy IA ; HMSN IA HMSN1A؛ CMT Autosonal Dominant With Focally Foded Myelin Sheatgs Type 1A شناخته میشود. این ژن در بازوی کوتاه کروموزم 17 قرار دارد و مسئول تولید پروتئین 22 میلین محیطی است. این پروتئین در سلولهای شوان ساخته میشود. وجود یک کپی اضافی از این ژن در مبتلایان باعث میشود فرایند پرازش این پروتئین به خوبی انجام نگیرد. وجود پروتئینهای ناقص باعث اختلال در جفت و جور شدن پروتئینهای سالم میشود. ساخت پروتئینهای میلین پردازش نشده فعالیت سلولهای شوان را مختل میکند که ناپایداری و یا نقصان کامل میلین را به همراه دارد. البته موتاسیونهای نقطهای در ژن PMP22 نیز رخ میدهد که تحت عنوان شارکوت ماری توث نوع 1E تقسیم بندی میشود. علاوه بر این حذف ژن PMP22 نیز باعث بروز بیماری palsies neuropathy with liability to pressure (HNPP) میشود. وجود یک الل طبیعی (مونوزومی) باعث بروز عوارض بالینی خفیف میشود. وجود دو الل طبیعی وضعیت طبیعی را در پی دارد. دوپلیکاسیون در یک نسخه از ژن (تریزومی) موجب بروز عوارض بالینی شدید بیماری میشود. دوپلیکاسیون در هر دو نسخه ژن (تترازومی) شدیدترین نشانههای بالینی بیماری را در پی خواهد داشت. در این آزمایشگاه بررسی دوپلیکاسیون و یا حذف این ژن با روش Real Time PCR Quantitative انجام میگیرد. در این روش تعیین کمیت قطعات DNA با اندازه گیری آستانه ورود به فاز لگاریتمی (Ct) در آزمایش PCR انجاممیشود. به این ترتیب تترازومی، تریزومی، منوزومی و یا نرمال بودن ژن PMP22 شناسایی میشود. البته یک کنترل داخی نیز مورد نیاز است که تفاوت در تکثیر آن با ژن PMP22 مقایسه میشود. این کنترل داخلی معمولاً ژن آلبومین در نظر گرفته میشود. با رسم نمودار تکثیر ژن مرجع و PMP22 وضعیت فرد مشخص میشود. در نمونههای نرمال ورود به فاز تکثیر برای هر دوی این ژنها تقریباً به صورت همزمان است. اما حذف یک کپی از این ژن باعث تأخیر ورود به فاز لگاریتمی میشود. وجود یک نسخه اضافی از ژن نیز باعث میشود ژن PMP22 زودتر از ژن آلبومین وارد فاز لگاریتمی شود. این آزمایش برای هر نمونه به صورت دوبلکس و یا تریپلکس انجام میگیرد. شارکوت ماری توث نوع 2A شارکوت ماری توث نوع 2A با نوروپاتی حسی و حرکتی اکسونهای اعصاب محیطی مشخص میشود. شدت بیماری در اندامهای پایینی مثل پا بیشتر از اندامهای بالایی مثل دست است. علائم اولیه بیماری در دهه اول یا دوم زندگی فرد ظاهر میشود. استفاده از ویلچر برای بعضی بیماران ضروری است اما اکثر این افراد نیازی به استفاده از این وسیله ندارند. جهش در ژن MFN2در ارتباط با اکثریت مبتلایان به شارکوت ماری توث نوع 2A شناسایی شده است. این بیماران را در زیر گروه CMT2A2 دسته بندی میکنند. تعدادی از مبتلایان به شارکوت ماری توث نوع دو A نیز تحت عنوان CMT2A1 دسته بندی میشوند. در این افراد ژن KIF1B جهش پیدا کرده است. تعداد اینگونه بیماران بسیار کم است. هر دو این بیماریها با الگوی توارث اتوزومی غالب به ارث میرسند. در این آزمایشگاه ژن MFN2 با روش توالی یابی مورد بررسی قرار میگیرد. این ژن روی بازوی کوتاه کروموزم یک قرار دارد و کد کننده پروتئین GTPase میتوفیوژن میتوکندریایی است. برای توالییابی ژن ابتدا با استفاده از پرایمرهای اختصاصی در آزمایش PCR تمام 17 اگزونهای ژن تکثیر میشود. محصول این PCR به همراه نمونه پرایمر فورارد مورد استفاده برای توالی یابی ارسال میشود. هر نمونه همراه با کنترل منفی و مثبت مورد آزمایش قرار میگیرد. با بررسی نتایج ارسالی وجود جهش در این ژن شناسایی میشود. شارکوت ماری توث نوع 4A شارکوت ماری توث نوع 4 گروهی از بیماریهای عصبی پیشرونده هستند که باعث نوروپاتی میلین زدا و اکسونی میشود. همانطور که گفته شد الگوی توارثی این گروه از بیماری شارکوت ماری توث اتوزومال مغلوب است. شایعترین زیر گروه شناخته شده این بیماری CMT4A است. تخمین زده میشود در حدود 25 درصد مبتلایان به شارکوت ماری توث نوع 4 جزء این زیر گروه باشند. تشخیص این بیماری با استفاده از یافتههای بالینی و انجام آزمایش ژنتیکی انجام میگیرد. تنها ژن شناخته شده در ارتباط با این بیماری GDAP1 است. این ژن روی بازوی بلند کروموزم 8 قرار دارد و جهشدر این ژن در مواردی با میلین زدایی و در مواردی نیز با نورپاتی اکسونی همراه است. CMT4A یکی از انواع شدید و پیش رونده شارکوت ماری توث است به طوریکه علائم اولیه بیماری معمولاً در نوزادی یا اوائل کودکی بروز میکند. در اکثر مواقع پیشروی بیماری باعث ناتوانی در دهه اول یا دوم زندگی فرد میشود و اکثریت مبتلایان ناچار به استفاده از ویلچر میشوند. مراحل پیشروی حتی در افراد مبتلا از یک خانواده نیز ممکن است تفاوت داشته باشد. در این آزمایشگاه تشخص مبتلایان به این بیماری با روش تعیین توالی انجام میگیرد. در این روش ابتدا هر 6 اگزون ژن GDAP1 با انجام PCR با پرایمر اختصاصی تکثیر مییابد. سپس محصول این PCR برای توالی یابی ارسال میشود. این آزمایشات برای تشخیص پیش از تولد در بارداریهای پرخطر نیز انجام میگیرد. در این موارد DNA استخراج شده در نمونههای جنیی که به روش آمینوسنتز و یا CVS گرفته شده مورد آزمایش قرار میگیرد. البته پیش از انجام این آزمایش الل بیماری زا باید در فرد بیمار خانواده شناسایی شود. |
| زمان جوابدهی: | 2الی 3 هفته پس از نمونه گیری |
مرکز ژنتیک پزشکی ابن سینای تبریز![]()
دوشنبه ۲۵ خرداد ۱۴۰۵